

Amyloidosis is considered a rare systemic disease, meaning it can affect different organs of the body, including the heart, kidneys, liver and nervous system (Papingiotis et al, 2021). Prevalence worldwide is considered to be highly under-recognised (Ruberg et al, 2019; Kittleson et al, 2020), but it has been on the rise since 2007 (Quock et al, 2018; Ney et al, 2023). Limited information is available about the prevalence of systemic amyloidosis, but in the UK, it is estimated to affect 8.0 per million inhabitants per year (Pinney et al, 2013). In the UK, there are at least 6000 cases at any time (Royal Free Charity, 2024), with imaging developments improving recognition and formal diagnosis, including of amyloidosis involving the heart (Maurer et al, 2017).
Over time, cardiac amyloidosis results in a restrictive cardiomyopathy, characterised by the stiffness of the heart's chambers, usually at an average age of 60-70 years (Wechelakar, 2021). Between first symptom onset and diagnosis confirmation there is at least a 1-year delay (Lousada et al, 2015), and prognosis worsens once cardiac involvement is recognised (Westin et al, 2022). The vast majority of cardiac amyloidosis is due to transthyretin amyloidosis (ATTR) and immunoglobulin light chain amyloidosis (AL). ATTR cardiomyopathy accounts for 13% of heart failure patients and 6-15% of aortic stenosis cases (González-López et al, 2015; Scully et al, 2018)
ATTR is predominantly found in Portugal, Japan, Sweden, and the USA. Some forms of cardiac amyloidosis are hereditary, and a subtype of ATTR cardiac amyloidosis almost exclusively affects individuals of African or Afro-Caribbean descent (Maurer et al 2017). For AL cardiac amyloidosis, little is known on its prevalence due to lack of multicentre studies (Bondermann et al, 2020; Ruiz-Hueso et al, 2023).
Early diagnosis of cardiac amyloidosis allows prompt initiation of treatments, reducing mortality and cardiovascular-related hospitalisations (Oerlemans et al, 2019). However, early and opportunistic diagnosis remains challenging, so raising awareness is the first step to improve patients' outcomes by reducing time to treatment. To enable nurses to understand and play a role in early recognition of cardiac amyloidosis, this article provides an overview of its pathophysiology, red flags and what to expect of its complex management (Walkowiak and Domaradzki, 2020).
Pathophysiology
Amyloidosis occurs when amyloid fibrils, which are aggregates of low molecular weight proteins, become insoluble and resistant to degradation. They then deposit in organs and connective tissues leading to disease (Wechalekar et al, 2016). The accumulation of these fibrils progressively leads to structural or functional damage of the involved organ, potentially leading to organ failure if left untreated.
Amyloidosis is classified according to the amyloid fibril protein involved and the classification terminology always starts with the letter ‘A’ (standing for amyloid), followed by an abbreviation of the protein name (Kittleson et al, 2020) (Table 1). There are more than 30 proteins capable of forming amyloid deposits in the human body, but only 9 can cause cardiac disease: Table 1 deals with the most commonly involved proteins. Cardiac amyloidosis occurs when the fibrils infiltrate the myocardium leading to disease of the muscle and stiffening of the heart's walls, referred to as restrictive cardiomyopathy (Benson et al, 2018). It can also lead to disorders of the electrical conductivity of the heart (cardiac arrhythmias), especially in the elderly (Ruberg et al, 2019).
Table 1. Amyloidosis classification
| Classification based on protein type | Hereditary | Most commonly affected organs |
|---|---|---|
| AL – Immunoglobulin light chain | Type affecting heart not hereditary | Kidneys and heart |
| AA – Serum amyloid A | No | Kidneys |
| AFib – Fibrinogen alpha chain variants | Yes | Kidneys |
| ATTRv – Transthyretin gene variantATTRwt–Transthyretin wild type | YesNo | Heart and peripheral nervous system |
The proteins affecting the heart and causing cardiac amyloidosis can be acquired throughout life or inherited (Garcia-Pavia et al, 2021). Over 98% of cardiac amyloid cases are due to AL or ATTR proteins, but these proteins can also affect other organs (Figure 1). When there is heart involvement the survival rates are quite poor, with a median survival of 2.5 years for ATTRv and 3.6 years for ATTRwt compared with approximately 8 years in patients without cardiac amyloidosis (Lane et al, 2019).
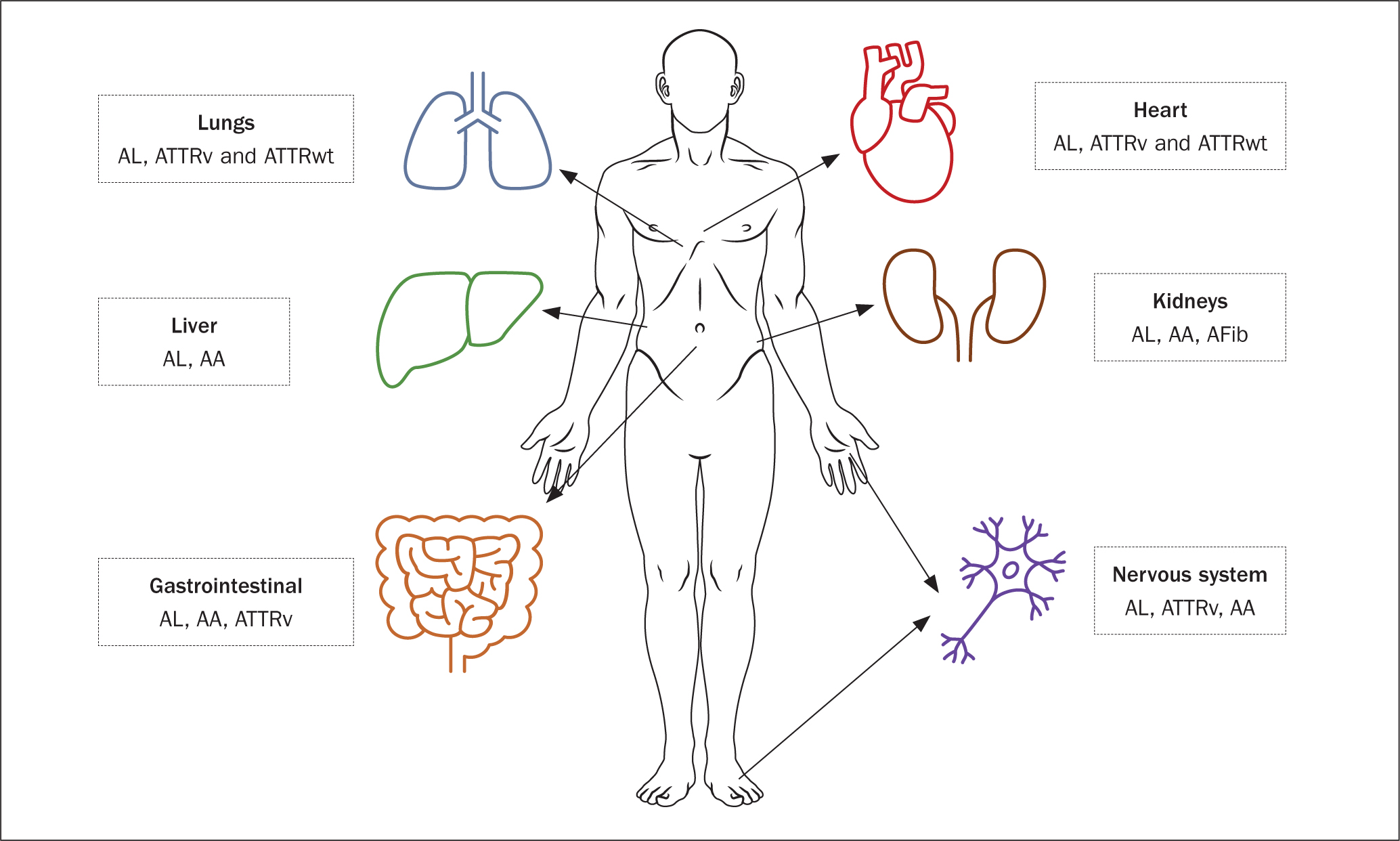
ATTR cardiac amyloidosis
Serum transthyretin (TTR) proteins are usually produced in the liver and responsible for carrying thyroid hormones (T4) and vitamin A (retinol). TTR can also be present in smaller amounts in the brain and eyes, synthesised by the choroid plexus (in the brain) and the retinal epithelium (in the eyes) (Minnella et al, 2021). Despite its role in thyroid function, there is currently no evidence of a link between thyroid disease and amyloidosis.
The hereditary form of ATTR (ATTRv) results from a genetic mutation that can cause changes in the structure of the TTR protein, causing it to misfold, aggregate and deposit in different organs and tissues (Papingiotis et al, 2021). It is passed on through generations and genetic testing is advisable for adult relatives of patients diagnosed with this condition.
The wild-type ATTR (ATTRwt) does not have genetic causes and is associated with the normal ageing process (previously named senile cardiac amyloidosis) with symptoms usually starting after 65 years of age (American Heart Association, 2021). It occurs as the TTR protein becomes prone to misfolding with increased age (Ruberg et al, 2019).
AL cardiac amyloidosis
A normal antibody (immunoglobulin) has two immunoglobulin heavy chains and two free light chain (kappa and lambda) proteins. In AL amyloidosis there is an overproduction of free immunoglobulin light chain by the bone marrow. These travel the bloodstream in abnormal levels and form amyloid deposits. The heart can be affected when misfolded light chains infiltrate the myocardium, causing problems with the integrity of the cells, potentially resulting in injury and death (Grogan et al, 2017). Abnormal and life-threatening heart rhythms such as pulseless electrical activity, ventricular fibrillation or bradycardias are also possible due to amyloid infiltration in the heart (Grogan and Dispenzieri, 2015).
Signs and symptoms
Most patients exhibit symptoms in the later stages of disease only and have already experienced a long history of malaise and weight loss (Clark et al, 2022). Signs or symptoms can vary depending on cardiac amyloidosis type (AL or ATTR), but the most common are depicted in Figure 2.
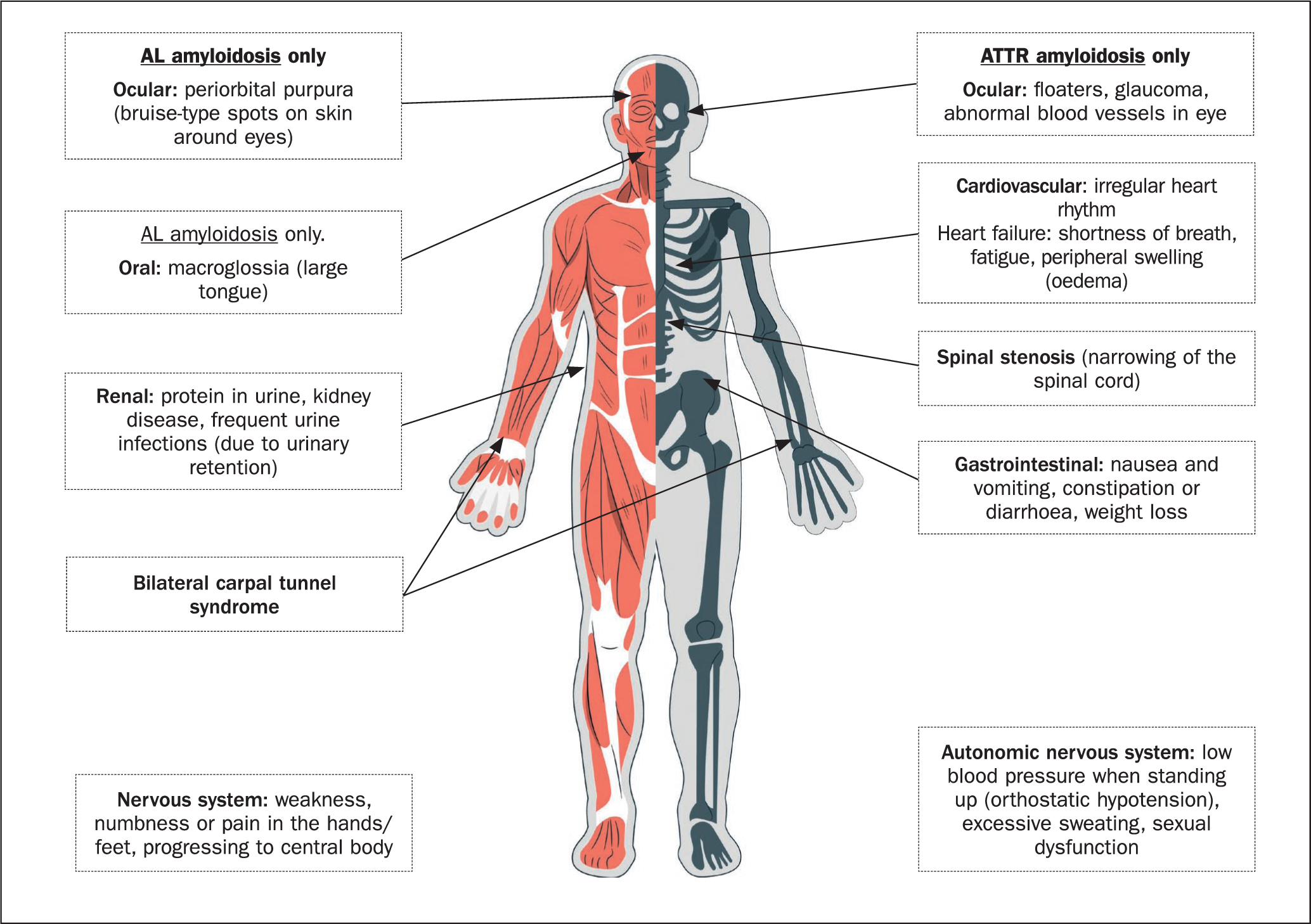
Produced in the body due to cardiac stress, brain natriuretic peptide (BNP) and N-terminal proBNP (NT-proBNP) are cardiac biomarkers, widely used in the diagnosis of heart failure and usually very elevated in cardiac amyloidosis. These biomarkers are collected via a blood test and, aside from being useful in detecting cardiac involvement in amyloidosis, they also have value in evaluating severity of disease depending on how elevated the levels are (Kyriacou et al, 2018).
The impact of cardiac amyloidosis on a patient's functional status and wellbeing continues to be assessed mainly by quantitative measures of quality of life. A deep and holistic understanding of how disease affects patients' daily lives is also required (Lane et al, 2019). Although qualitative accounts are scarce, one study exploring ATTR patients' experiences with amyloidosis highlighted negative effects across physical, mental and social wellbeing. A sense of loss of autonomy and inability to perform daily tasks shows this to be a highly complex and multifaceted disease (Lovley et al, 2021).
Diagnosis
Because amyloidosis can affect multiple organs, the signs and symptoms are varied and can be mistaken for other common diseases. This mixed presentation makes diagnosis very hard and often delayed; it normally includes a suspicious phase, based on red flags (Table 2), and a diagnosis phase, where investigations are made and type confirmed (Table 3) (Garcia-Pavia et al, 2021). In a survey of patients with ATTR amyloidosis, many saw more than five clinicians before being diagnosed correctly (Lousada et al, 2015). Early identification is key for better mortality, morbidity and quality of life, allowing newly available treatments to be offered in the early stages of the disease (Gertz et al, 2020).
Table 2. Common red flags in amyloidosis
| Cardiac |
|
| Peripheral nervous system |
|
| Laboratory |
|
| Cardiac imaging findings |
|
Table 3. Example of tests used to diagnose cardiac amyloidosis
| Cardiac amyloidosis type | Non-invasive and invasive tests |
|---|---|
| AL and ATTR | Blood test for serum free light chain concentration+ Blood and urine test for specific proteins called ‘immunofixation’+ ECG, echocardiogram and/or cardiac magnetic resonance (CMRi)+ Blood test for NT-proBNP and troponin levels |
| AL | All of the above and:+ Bone marrow biopsy or+ Heart biopsy |
| ATTR | All of the above and:+ Scintigraphy, a scan creating images of organs and tissues using radioactive material (radiotracers) |
Advances in technology and more accurate cardiac imaging techniques have improved the diagnosis of amyloidosis. Despite still being considered a rare disease, in the 2010s a higher number of patients was being referred to the National Amyloidosis Centre in London (Lane et al, 2019)
Clinical management
The main focus in cardiac amyloidosis is to treat and prevent complications such as heart failure and arrhythmias (supportive treatment), and to halt the progression of amyloid deposition (disease-modifying) (Papingiotis et al, 2021). These are illustrated in Table 4. Heart failure therapies should be considered when the left ventricular function is reduced, commonly referred to as heart failure with reduced ejection fraction (HFrEF). Unfortunately, these medications are often poorly tolerated due to symptomatic hypotension (McDonagh et al, 2021). Complicating matters further, currently there is no evidence of an optimal follow-up scheme for those with cardiac amyloidosis. Patients are commonly seen at 6-month intervals for an electrocardiogram (ECG) and blood tests (including renal profile, NT-proBNP and troponin), and once a year have an echocardiogram and 24-hour ECG (Garcia-Pavia et al, 2021).
Table 4. Treatment and management of cardiac amyloidosis
| Disease modifying | |
| AL amyloidosis | Management similar to cancer, with chemotherapy and or stem-cell transplantation to target the plasma-cell causing the AL amyloid formation |
| ATTRwt and ATTRv amyloidosis | Tafamidis is an oral medication that reduces the formation of TTR amyloid. It is not currently given in the NHS as it is not deemed to be cost-effective by the National Institute for Health and Care Excellence (NICE) (2021) |
| ATTRv amyloidosis |
|
| Supportive treatment | |
| Heart failure (HF) |
|
| Arrhythmias |
|
Patients are often very symptomatic and will require specialist input for co-ordination of care, titration of diuretics, monitoring and support with self-care, usually through heart failure specialist nurses (National Institute for Health and Care Excellence (NICE), 2018). The patient's quality of life is often reduced owing to the significant changes to their level of physical activity and activities of daily living (Aimo et al, 2021). Emotional support is essential as significant levels of anxiety and depression are commonly found in this population (Smorti et al, 2016). To improve satisfaction with life and care, a multidisciplinary team (MDT) approach involving different specialties and areas of care is needed (Koike et al, 2021; Sperry et al, 2022), with access to occupational therapy, physiotherapy, psychological support and social care. Palliative care involvement is necessary and conversations about advanced care planning decisions should start early on the patient's trajectory (NICE, 2018) due to the progressive life-threatening nature of this disease.
Conclusion
Cardiac amyloidosis is a challenging journey for patients, with diagnosis being complex and delayed due to its heterogeneous presentation. Recent research and advances in imaging techniques have improved its recognition, but regular screening and increased awareness among health professionals are fundamental to improve disease prognosis and time to treatment. Morbidity remains high and symptom support is key due to lack of curable options. Increasing knowledge and awareness will enable nurses to empower patients on managing their progressive and life-threatening disease trajectory. Encouraging and supporting patients to self-care, including identification of signs and symptoms of deterioration and heart failure, is important to avoid hospital admissions due to fluid overload. Working collaboratively with members of the MDT is essential to improve patient satisfaction with care and quality of life.
KEY POINTS
- Amyloidosis is a rare but increasingly prevalent disease that can affect multiple organs. Cardiac involvement worsens prognosis and should be suspected for patients presenting with signs and symptoms of heart failure but otherwise normal left ventricular function
- Anyone involved in direct patient care can look out for the red flags of cardiac amyloidosis. There is at least 1 year's delay between symptoms emerging and formal diagnosis – awareness is key
- Management is mostly focused on controlling symptoms and maintaining quality of life. Medications often used in other cardiac diseases, including heart failure, are often not tolerated due to hypotension and bradycardia
CPD reflective questions
- What are the challenges in the diagnosis of cardiac amyloidosis and how can health professionals improve early identification?
- Cardiac amyloid patients can present with signs and symptoms of heart failure. How can you support these patients in coping with physical and psychosocial challenges?
- What are the red flags for cardiac amyloidosis? How can you advocate for the inclusion of screening and education initiatives within your workplace to improve early detection?